深研院潘锋团队在发展图论结构电化学与AI相融合应用于尿素电催化机理研究取得进展

电催化是实现可持续能源转化、减碳减排的关键技术。对电催化反应路径的认识是合理设计催化剂的前提,受限于复杂的表面动态效应和庞大的反应网络,这需要发展新的研究范式来实现催化反应路径的高效预测。北京大学深圳研究生院新材料学院潘锋教授团队将数学的图论和结构化学相融合,把材料中的原子当作图论的点,原子间的化学键当作图论的边,创新性地提出一种基于图论的结构化学研究方法(Sci China Chem, 2019, DOI: 10.1007/s11426-019-9502-5)解决了晶体学同构判断的难题,建立了拥有65万晶体结构的大数据系统,并以此发展材料基因组学和AI for Science(AI4S)的研究,将其应用于低维材料的发现(National Science Review, 2022, DOI: 10.1093/nsr/nwac028)和新型固态电解质的设计(J. Am. Chem. Soc. 2024, 146, 27, 18535-18543)。
近日,潘锋团队将该方法进一步扩展用于电化学催化反应研究,发展了图论结构电化学,诠释了电催化尿素合成反应的机理,结合AI发展了基于图表示、图同构和机器学习的主动学习框架,可以实现从包含数百个中间物种反应网络中快速预测最优热力学路径的方法。该方法可以显著减小全局探索反应路径所需的计算量,为高通量设计新型催化剂提供一条新途径。相关研究成果以“Automating discovery of electrochemical urea synthesis reaction paths via active learning and graph theory”为题,发表在中国化学会旗舰期刊CCS Chemistry 2024, 7, 1-14。
涉及多电子转移的电化学反应通常具有一个庞大的反应网络特征,该网络包含了大量基元反应。探索最可能的反应路径是催化机制分析中的核心任务。量子化学方法,如密度泛函理论(DFT),常被用来阐明反应机制。然而,由于化学直觉的局限性和计算需求,这些方法通常仅适用于简单的反应过程,在处理具有非均匀结构或组成的复杂反应网络时会遇到困难。这一问题在二氧化碳电还原为多碳产物和电化学C-N偶联等蓬勃发展且广受关注的领域中尤为重要。为降低计算成本,研究者们已开发了多种AI机器学习算法并取得进展,但其中大多数仅在静态、完整的金属和合金表面以及典型反应上进行了验证。值得注意的是,当前大部分机器学习替代模型中忽略了催化剂表面重构的动态效应。然而,在研究涉及大分子、高覆盖率吸附物和活性表面的电化学反应时,表面动态效应尤为重要。在该情景下,AI机器学习替代模型的核心挑战在于难以准确评估重构表面和吸附物的能量,进而难以实现最优路径的预测。
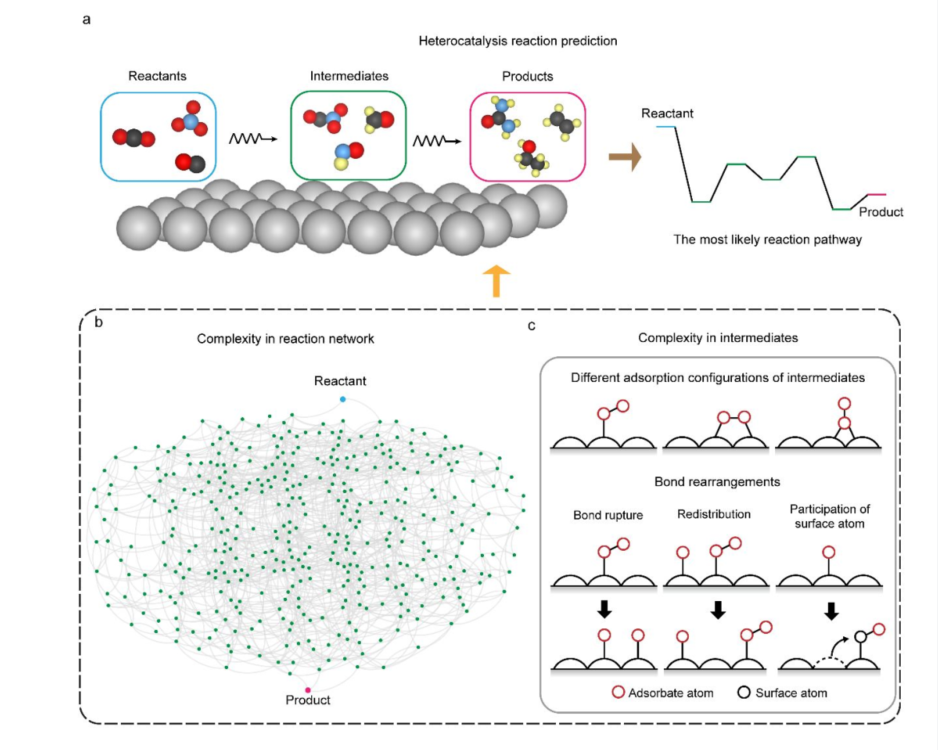
电催化剂反应网络复杂性及动态重构示意图
潘锋团队引入了一种结合图论和AI主动学习循环的工作流程,以解决复杂表面电催化反应路径预测这一长期存在的挑战。通过图论方法,以尿素电化学催化合成为例,遍历了其催化合成反应中大多数键重排类型及相应的重构模式,通过AI机器学习方法对反应网络中间体稳定性和形成能进行预测,降低了密度泛函理论的计算成本。以在氮掺杂石墨烯作为催化剂为例,该图论结构电化学理论在尿素电催化合成这一反应中得到验证,说明该理论框架的有效性。该反应体系在工作条件下发生显著动态结构重构。整个反应网络包含901个反应物种,利用该框架,只需计算其中的40%就可实现对反应网络能量学的评估并得到过电势。此图论结构电化学框架可以扩展到其他复杂的电化学反应,并在最小程度依赖精确量子化学计算的前提下,促进过电位的快速估算,从而为真实条件下催化机制的自动化计算分析铺平道路。
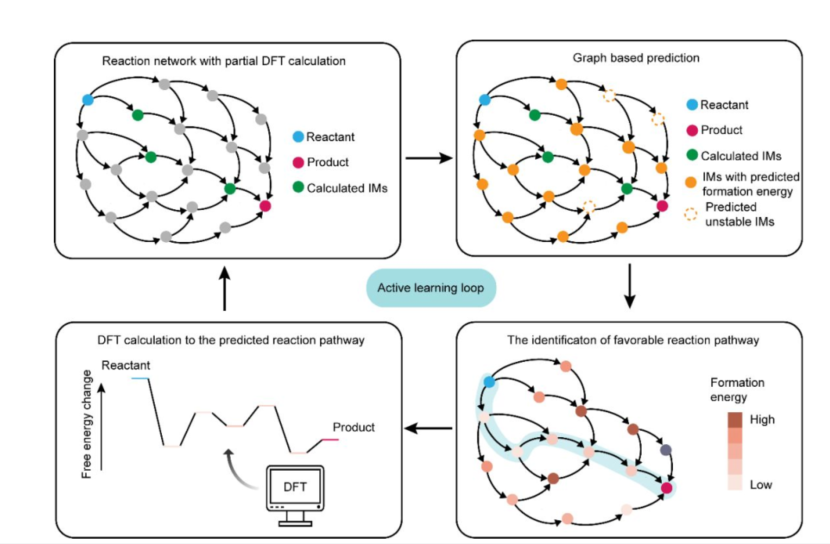
主动学习流程概述
潘锋、北京大学深圳研究生院新材料学院李舜宁副研究员为本文通讯作者。北京大学深圳研究生院新材料学院博士生毕业生、现任厦门大学特任副研究员郑世胜为本文第一作者。论文得到国家自然科学基金、广东省和深圳市相关平台支持。





